Fragen und Anworten
Die DBA wird durch eine Störung der Funktion der Ribosomen in den Zellen verursacht. Die Aufgabe von Ribosomen ist es, Proteine (Eiweiße) her zu stellen, indem sie Aminosäuren (Eiweißbausteine) aneinanderfügen. Als Bauanleitung oder "Kopiervorlage" bei der Bildung der unterschiedlichen Proteine im Ribosom dient die Boten-Ribonukleinsäure (enthält die genetische Information der DNA, auch messenger-RNA, mRNA genannt). Diese "läuft“ durch das Ribosom, wenn Proteine gebildet werden sollen. So können die Aminosäuren in die richtige Reihenfolge gebracht und miteinander verbunden werden. Die so entstandene Aminosäurenkette (das fertige Protein) verlässt dann das Ribosom, um seine jeweiligen Aufgaben zu verrichten (siehe Abbildung 1).
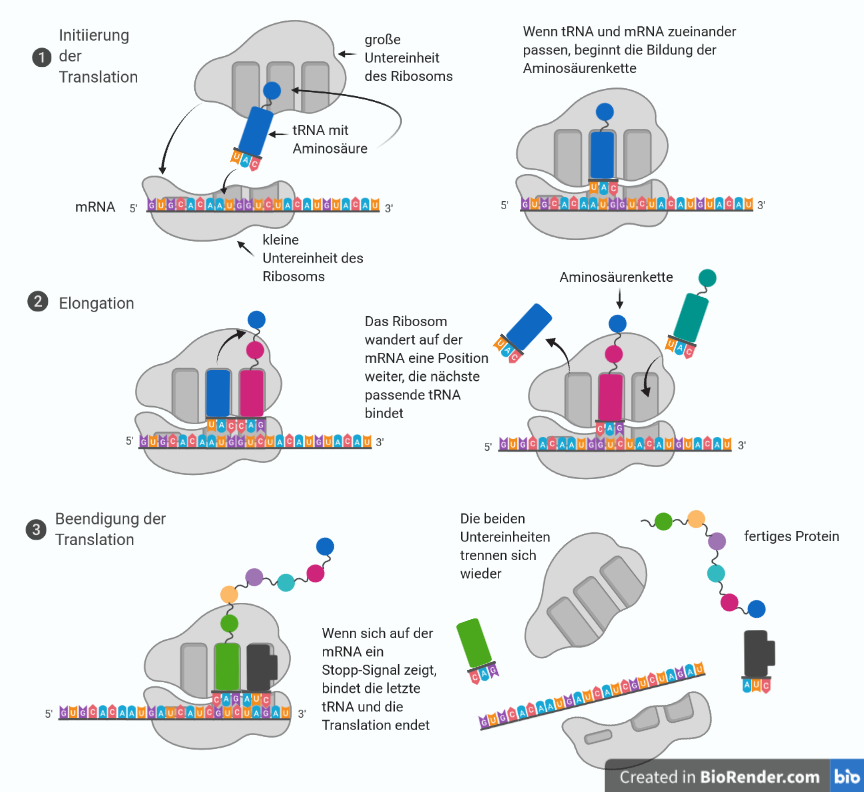
Abbildung 1: Ribosom bei der Arbeit
Ribosomen bestehen aus einer großen und einer kleinen Untereinheit. Sie setzen sich selbst aus Proteinen und Ribonukleinsäure (RNA) zusammen. Ursache der DBA sind unterschiedliche Mutationen (Genveränderungen), die überwiegend zu einer fehlerhaften Zusammensetzung der Ribosomen, d.h. ihrer Proteine oder Ribonukleinsäuren, führen.
Die Störung der Proteinproduktion in den Ribosomen hat wichtige Konsequenzen, da Proteine viele Aufgaben bewerkstelligen, die ein Organismus zum Leben braucht. Alle kernhaltigen Zellen des Menschen, zu denen auch die blutbildenden Zellen im Knochenmark und die Knochenzellen gehören, bilden und enthalten Proteine und verständigen sich untereinander durch sie. Ebenso transportieren Proteine lebenswichtige Substanzen, beispielsweise transportiert das Hämoglobin (roter Blutfarbstoff) den Sauerstoff im Körper und bringt ihn zu den Organen. Antikörper sind ebenfalls Proteine, die Fremdes markieren und so zur körpereigenen Abwehr beitragen. Neben diesen gibt es noch zahlreiche andere Proteine mit vielen verschiedenen Funktionen, die für einen gesunden Organismus unverzichtbar sind. Da die DBA bei einem betroffenen Patienten auch schon während der frühen Entwicklung im Mutterleib vorhanden ist, lässt sich das Auftreten von angeborenen Fehlbildungen gut erklären.
Warum die DBA sich in erster Linie im Knochenmark mit einer Störung der roten Blutbildung zeigt und warum die Ausprägung der Symptome bei einzelnen DBA-Patient*innen so unterschiedlich ausfällt oder auch einmal Symptome fehlen, ist noch nicht verstanden.
Viele Mutationen führen zu einer fehlerhaften Zusammensetzung und Funktion der Bestandteile der großen oder der kleinen Untereinheit der Ribosomen oder anderer für die Funktion der Ribosomen wichtiger Strukturen.
Bei ca. 70-80% der DBA Patient*innen können für die Erkrankung ursächliche Mutationen nachgewiesen werden. Sie betreffen die Proteine der großen Untereinheit (ribosomal protein large, RPL) oder der kleinen Untereinheit (ribosoma protein small, RPL) der Ribosomen oder seltener andere für die Funktion der Ribosomen wichtige Strukturen. Die Mutationen bestehen meistens aus dem Austausch einer oder weniger Nukleinsäuren in dem betroffenen Gen. Insgesamt sind hier aktuell 28 Mutationen in ribosomalen Genen bekannt. Bei wenigen DBA-Patient*innen fehlen ganze Abschnitte eines Chromosoms mit mehreren verschiedenen Genen (dies nennt man auch Deletion), wobei eines der dann fehlenden Gene für ein Protein der Ribosomen kodiert. Mutationen in den Genen für die Proteine RPS19, RPL5, RPS26 und RPL11 sind für ca. 70% -80% aller bekannten Genveränderungen bei DBA verantwortlich (s. Abbildung 2).
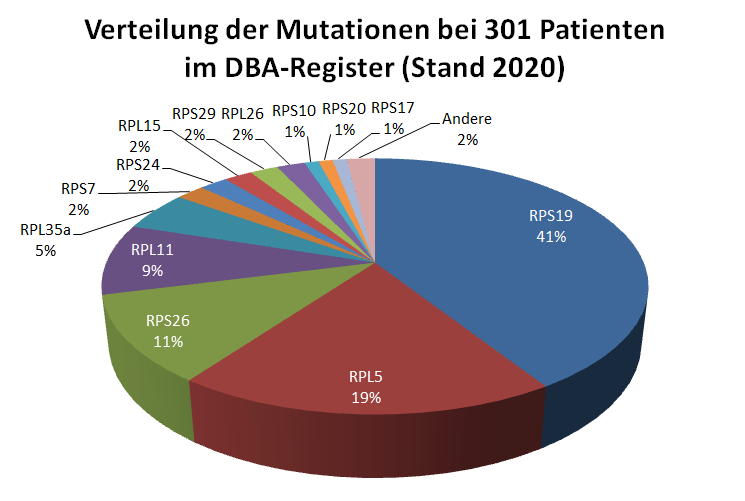
Abb 2. Verteilung der Mutationen
Die Mutation, die zu einer DBA führt, kann entweder von einem Elternteil vererbt sein oder in einem frühen Stadium des Embryos des betroffenen DBA-Patienten neu entstehen. Ursachen oder Auslöser für das Auftreten solcher neuen Mutationen kennt man wie bei vielen anderen Erkrankungen noch nicht.
Die Mutationen betreffen immer alle Körperzellen und können an die Nachkommen vererbt werden. Die Wahrscheinlichkeit, eine DBA-Mutation an die nächste Generation zu vererben, beträgt 50%. Diese Vererbungsform nennt man autosomal-dominant, da die Vererbung einer DBA-Mutation von nur einem Elternteil ausreicht, um die Erkrankung bei einem Nachkommen auszulösen.
Sind in einer Familie mehrere Mitglieder von DBA betroffen, haben alle die gleiche ursächliche Mutation. Die Symptome der DBA der betroffenen Familienmitglieder können aber sehr unterschiedlich sein und reichen von keinen gesundheitlichen Problemen bis zur Transfusionsbedürftigkeit. Der Grund für die unterschiedliche Ausprägung der Erkrankung ist weitgehend unbekannt. Auch wenn im Allgemeinen die Ausprägung der DBA von Generation zu Generation zunimmt (Antizipation), ist eine Vorhersage zur Schwere der Krankheit eines Trägers einer DBA-typischen Mutation nicht möglich.
Allen Patient*innen mit DBA sollte eine humangenetische Beratung angeboten werden. Eine Pränataldiagnostik (genetische Diagnostik vor der Geburt) und eine Präimplantationsdiagnostik (im Falle einer in vitro-Fertilisation/künstlichen Befruchtung) für das Vorliegen einer bei einem Elternteil bekannten DBA-Mutation sind möglich, wobei der Schweregrad der Ausprägung der Erkrankung allerdings nicht vorausgesagt werden kann.
Die gesundheitlichen Probleme von Patient*innen mit einer DBA sind im Wesentlichen durch eine Störung der Blutbildung im Knochenmark und deren notwendiger Behandlung bedingt. Eine verringerte Produktion von Erythrozyten führt zur Anämie, seltener kommt es auch zu einer Verminderung der Produktion von Leukozyten (weißen Blutkörperchen) und/oder Thrombozyten (Blutplättchen). Begleitende Fehlbildungen können operative Korrekturen notwendig machen. Patient*innen mit einer DBA haben ein erhöhtes Krebsrisiko. Die Höhe dieses Risikos und seine Bedeutung sind zurzeit noch nicht eindeutig zu benennen. Organschäden können infolge der Behandlung der Anämie mittels Bluttransfusionen entstehen. Besonders in der Leber, der Bauchspeicheldrüse (Pankreas) und im Herzmuskel sind Schäden durch eine Eisenüberladung als Folge der Bluttransfusionen möglich, auch bei nicht-transfundierten Patient*innen kann es zu einer Eisenüberladung kommen.
Das Blutsystem
Im Knochenmark werden Erythrozyten, Leukozyten und Thrombozyten gebildet. Bei einer DBA kann es grundsätzlich zu einer verringerten Produktion aller drei Zellreihen kommen (Panzytopenie). In fast allen Fällen steht dabei die verminderte Bildung von Erythrozyten mit der hieraus folgenden Anämie im Vordergrund. Die Leukozytenzahl im Blut ist bei vielen DBA-Patient*innen leicht erniedrigt, ohne dass dies gesundheitliche Auswirkungen hat.
Typische Beschwerden von Patient*innen mit Anämie sind zunehmende Blässe, Herzrasen, Kopfschmerzen, rasche Ermüdbarkeit, Luftnot bei körperlicher Anstrengung, bei Kleinkindern Trinkunlust und Gedeihstörung. Auch wenn insbesondere kleine Kinder bei deutlicher Anämie oft wenig beeinträchtigt erscheinen, ist davon auszugehen, dass eine Anämie sich langfristig negativ auf den Gesundheitszustand eines DBA-Patienten auswirkt. Daher wird eine konsequente Behandlung der Anämie bei DBA-Patient*innen in jedem Lebensalter empfohlen.
Mögliche Fehlbildungen
Etwa 40% der DBA-Patient*innen haben angeborene Fehlbildungen, die unterschiedliche Organe betreffen können. Häufige Fehlbildungen betreffen das Gesicht, wie z.B. eine Gaumenspalte, oder die Arme und den Daumen. Auch Herzfehler, Fehlbildungen an der Niere, Leistenhernien, ein angeborener grüner oder grauer Star, Hautveränderungen wie große „Leberflecke“ und Entwicklungsstörungen können auftreten. Die Körperlänge der meisten DBA-Patient*innen liegt im unteren Normbereich, einige Patient*innen haben einen Kleinwuchs.
Bei Kleinkindern mit DBA wurde ein typisches "DBA-Gesicht" mit Stupsnase, breitem Augenabstand, kräftiger Oberlippe, tieferliegenden Ohren und aufgewecktem Gesichtsausdruck beschrieben. Diese Auffälligkeiten scheinen aber mit dem Wachstum eher zu verschwinden.
Zum Krebsrisiko
DBA-Patient*innen haben gegenüber gesunden Gleichaltrigen ein erhöhtes Risiko im Laufe ihres Lebens eine Krebserkrankung zu entwickeln. Wie hoch diese Wahrscheinlichkeit ist, lässt sich zurzeit nur ungenügend abschätzen. Im Gegensatz zu anderen angeborenen Störungen des Knochenmarks (z. B. Fanconi-Anämie, schwere kongenitale Neutropenie, Shwachman-Diamond-Syndrom) ist das Risiko für das Auftreten einer Leukämie oder eines myelodysplastischen Syndroms (MDS) nach neueren Daten nur geringfügig oder nicht erhöht. Hingegen wurden in den letzten Jahren vermehrt Erkrankungen wie Brustkrebs, Darmkrebs oder andere Karzinome im jungen und mittleren Erwachsenenalter bei DBA-Patient*innen beobachtet.
Die Datenlage ist augenblicklich noch nicht gut genug, um für DBA-Patient*innen eine allgemeine Empfehlung im Sinne der Notwendigkeit eines frühzeitigen Vorsorgeprogramms zu entwickeln. Jährliche Gesundheitskontrollen beim Haus- oder Facharzt, Wahrnehmung der allgemeinen Vorsorgeangebote und Aufsuchen eines Arztes bei Beschwerden (Durchfälle, Blut im Stuhl, Schmerzen in der Brust, veränderte Hautflecken etc.) ist aber allen DBA- Patient*innen besonders im Erwachsenenalter ausdrücklich zu empfehlen. Das vermehrte Auftreten von Knochentumoren (Osteosarkome) bei Kindern, Jugendlichen und jungen Erwachsenen ist durch Fallberichte belegt, eine Früherkennung dieser Krebserkrankungen ist zurzeit nicht möglich.
Organschäden durch Eisenüberladung
Erythrozyten sind die Sauerstoffträger im Blut. Sie enthalten Hämoglobin, das über ein Eisenmolekül den Sauerstoff bindet. Bei einer Anämie versucht der Körper, die Produktion der Erythrozyten zu steigern. Dazu steigert er u.a. die Aufnahme von Eisen aus der Nahrung im Darm. Patient*innen mit DBA können dieses zusätzlich aufgenommene Eisen jedoch nicht verwerten, da die Blutbildung aufgrund der Funktionsstörung der Ribosomen gestört ist. Da der menschliche Körper keinen Mechanismus besitzt, das überschüssige Eisen auszuscheiden, entwickeln Patient*innen mit DBA eine Eisenüberladung. Das Eisen kann nicht verwertet werden und lagert sich in den Organen ab. Bluttransfusionen, die aus Erythrozyten bestehen, verstärken die Eisenüberladung, da Erythrozyten viel Eisen enthalten.
Durch die Eisenüberladung kommt es langfristig zu einer Schädigung vieler Organe, vor allem von Herz, Leber und verschiedenen Hormon-produzierender Drüsen (z.B. Schilddrüse, Hirnanhangs- und Bauchspeicheldrüse). Ohne eine adäquate Behandlung können daher unter Umständen schwere, manchmal auch tödlich verlaufenden Komplikationen auftreten wie Herzmuskelschwäche und Herzrhythmusstörungen (Kardiomyopathie), Leberentzündungen und –funktionsstörungen (Hepatopathie), Zuckerkrankheit (Diabetes mellitus), Kleinwuchs, verzögerte Pubertätsentwicklung, Schilddrüsenunterfunktion und Störungen des Vitamin D-Stoffwechsels.
DBA-Patient*innen, bei denen eine Eisenüberladung besteht und/oder Patient*innen, die regelmäßig Bluttransfusionen erhalten, werden daher mit Medikamenten behandelt, die das überschüssige Eisen an sich binden, so dass es über den Stuhl und den Urin ausgeschieden werden kann.
Wenn bei einem Patienten das Vorliegen einer DBA vermutet wird, ist zuerst die genaue Kenntnis der Krankheitsgeschichte (Anamnese) und die körperliche Untersuchung wichtig. Dann wird mit einer Blutentnahme das Blutbild überprüft. Durch die Bestimmung der Konzentration des Hämoglobins und der mikroskopischen Untersuchung des Blutausstrichs kann festgestellt werden, ob eine Anämie besteht, wie stark diese ausgeprägt ist und ob es sich vermutlich um eine DBA oder um eine andere Art von Anämie handelt.
Folgende Untersuchungen werden bei Verdacht auf DBA empfohlen:
- Erhebung der eigenen Krankengeschichte und der Familienkrankengeschichte (Anamnese, Familienanamnese)
Erklärung: Eine positive Familiengeschichte für eine DBA oder unerklärte Blutarmut im Kindesalter können auf eine DBA hinweisen.
- körperliche Untersuchung
Erklärung: Fehlbildungen können auf das Vorliegen einer DBA hinweisen.
- Bestimmung des Blutbilds mit mikroskopischer Differenzierung und Messung der Zahl der Retikulozyten
Erklärung: Vor der Geburt werden Erythrozyten gebildet, die andere Eigenschaften haben und aus anderen Bestandteilen bestehen als Erythrozyten, die nach der Geburt gebildet werden. Die Erythrozyten der Fetalzeit haben ein größeres Volumen (mittleres korpuskuläres Volumen, MCV), einen hohen Anteil an fetalem Hämoglobin (HbF) und eine erhöhte Konzentration des Enzyms ADA (siehe unten). Erythrozyten von DBA-Patient*innen weisen oft ähnliche Eigenschaften auf wie in der Fetalzeit.
- Basislabordiagnostik mit Leber- und Nierenwerten, Hämolyseparametern (Laborparameter, die bei einem Zerfall von Blutkörperchen erhöht sind), Ferritin (Maß für den Eisenhaushalt)
- Messung der Konzentration von Hämoglobin F (HbF) im Blut (möglichst vor Transfusion)
Erklärung: Während der Entwicklung des Menschen im Mutterleib werden unterschiedliche Hämoglobine gebildet. Kurz vor der Geburt besteht der Hauptanteil des Hämoglobins aus Hämoglobin F (fetales Hämoglobin, HbF), das sich von dem später gebildeten Hämoglobin A (adultes Hämoglobin) unterscheidet. Bei einer DBA werden vermehrt Erythrozyten gebildet, die HbF enthalten.
- Messung der Enzymaktivität der Adenosindeaminase (ADA) in Erythrozyten (möglichst vor der Transfusion)
Erklärung: Das Enzym ADA ist spielt eine wichtige Rolle im Stoffwechsel der Desoxyribonukleinsäure (DNA). Vor der Geburt finden sich hohe ADA-Konzentrationen in Erythrozyten. Die Erythrozyten von DBA-Patient*innen weisen in der Regel noch Eigenschaften auf, die sonst nur vor der Geburt gesehen werden. Hierzu gehört neben einem erhöhten HbF eine erhöhte ADA-Konzentration. Da durch eine Bluttransfusion die Konzentration der vom eigenen Körper gebildeten Erythrozyten verringert wird, sollte die Bestimmung von HbF und ADA möglichst vor einer Transfusion oder im großen Abstand zu dieser durchgeführt werden.
- Knochenmarkpunktion zur
- morphologischen Beurteilung der Knochenmarkausstriche (s. Abbildung 3)
- Durchführung einer Chromosomenanalyse in Knochenmarkzellen
- Ausschluss einer Parvovirus B19-Infektion im Knochenmark mittels der Methode einer Polymerasekettenreaktion (PCR)
Erklärung: Bei allen DBA-Patient*innen (auch nach vorausgegangener genetischer Sicherung der Diagnose) wird eine Untersuchung des Knochenmarks empfohlen. Die morphologische Betrachtung des Knochenmarks muss kompatibel mit der Diagnose DBA sein (Verminderung der Zahl der Vorstufen roter Blutkörperchen) und dient (wie die Chromosomenanalyse) als Ausgangspunkt für möglicherweise notwendige Untersuchungen zu einem späteren Zeitpunkt. Eine Infektion mit dem Parvovirus B19 (Erreger von Ringelröteln) kann wie die DBA zu einer hypoplastischen Anämie führen und ist nur durch eine negative PCR im Knochenmark sicher auszuschließen.
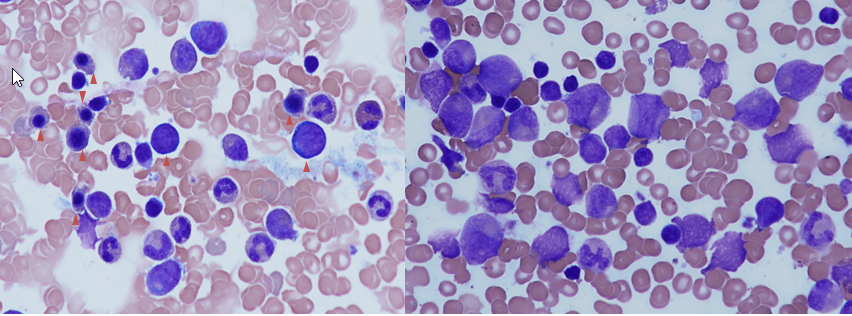
Abbildung 3: Knochenmarkausstriche von gesundem Patient (links) + DBA-Patient (rechts) im Vergleich. Die Zellen der roten Blutbildung (Pfeile) fehlen beim DBA-Patienten komplett.
- Molekulargenetische Diagnostik aller DBA-assoziierten Gene
Erklärung: Mittlerweile kann bei etwa 70-80% der Patient*innen eine krankheitsverursachende genetische Veränderung (Mutation oder Deletion) nachgewiesen werden. Bei Nachweis einer krankheitsverursachenden genetischen Veränderung ist die Diagnose DBA gesichert. Ist keine krankheitsverursachende genetische Veränderung nachweisbar, ist die Diagnose DBA jedoch nicht ausgeschlossen. Es ist möglich, dass bei der molekulargenetischen Diagnostik unklare Ergebnisse (Varianten unklarer Varianz) oder unerwartete Ergebnisse (Zufallsbefunde) gefunden werden. Daher ist vor der Untersuchung eine ausführliche Aufklärung erforderlich.
Ist die Diagnose DBA gesichert, sind folgende zusätzliche Untersuchungen empfohlen:
- Untersuchung zur Detektion nicht sichtbarer Fehlbildungen: Ultraschall des Herzens und des Bauches
- Bestimmung der Immunoglobulinwerte und Durchführung einer Lymphozytenphänotypisierung
- Bestimmung der Blutgruppe mit Antikörpersuchtest und direktem Coombs-Test
Wird bei einem Kind oder einem Erwachsenen eine DBA festgestellt, sollte die weitere Betreuung an einem spezialisierten Behandlungszentrum für Blutkrankheiten stattfinden. Das DBA-Register bietet zudem jederzeit ein persönliches Beratungsgespräch für Patient*innen und deren Familien an.
In spezialisierten Behandlungszentren hat das medizinische Personal Erfahrungen mit der Behandlung von Patient*innen mit seltenen Blutkrankheiten wie einer DBA. Die regelmäßige Vorstellung des Patienten in einem solchen Zentrum ist wichtig, damit der Krankheitsverlauf sorgfältig überwacht und Komplikationen frühzeitig erkannt und behandelt werden können. Außerdem können Fragen zum Umgang mit dem erkrankten Kind, zu den gängigen und neuen Behandlungsmethoden und aktuellen Forschungsergebnissen fachgerecht beantwortet werden. Allen DBA-Patient*innen sollte eine sozialmedizinische Beratung und psychologische Betreuung angeboten werden.
Die Art der Behandlung einer DBA richtet sich nach der Schwere der Krankheitszeichen und danach, ob ein Ansprechen auf eine bestimmte Therapieform beobachtet werden kann. Während viele DBA-Patient*innen durchgehend eine Behandlung benötigen, können einige Patient*innen phasenweise oder langfristig ohne Therapie auskommen. Grundsätzlich stehen folgende Behandlungsoptionen zur Verfügung:
- regelmäßige Bluttransfusionen (=Erythrozytenkonzentrate)
- Einnahme von Steroiden (=Cortison, Glukokortikoide)
- allogene Stammzelltransplantation (SZT)
- Gentherapie (in baldiger Zukunft)
- bisher für eine Behandlungsempfehlung durch Studien nicht ausreichend belegte, individuelle, experimentelle Therapieversuche
- Behandlung von Fehlbildungen
- Behandlung von Therapiefolgen, vor allem Eisenentzugstherapie
Bluttransfusion
Unter einer Bluttransfusion versteht man die Verabreichung von Erythrozyten von einem Blutspender. Patient*innen erhalten dabei über einen Venenzugang (meist in der Ellenbeuge) Erythrozyten von gesunden Spendern. Dabei werden in der Regel 10 – 15 ml Erythrozytenkonzentrat pro kg Körpergewicht transfundiert (ein Beutel Erythrozytenkonzentrat hat ein Volumen von ca. 250 – 300 ml). Transfusionsbedürftige Patient*innen mit DBA erhalten üblicherweise alle drei bis sechs Wochen eine Bluttransfusion. Bei Patient*innen, die regelmäßig Transfusionen benötigen, sollten die Transfusionen in einem festen Intervall und mit einem festgelegten Hämoglobinwert vor Transfusion (Ziel-Tal-Hämoglobinspiegel) erfolgen. Bei Kindern mit DBA wird empfohlen, diesen Hämoglobin-Talwert vor Transfusion mit 9,5 - 10 g/dl festzulegen. So können optimale Bedingungen zur ausreichenden Sauerstoffversorgung des Organismus, für eine altersgerechte Entwicklung der Kinder und Jugendlichen und eine bestmögliche Lebensqualität im Erwachsenenalter geschaffen werden. Nicht alle Patient*innen mit einer DBA bleiben ihr Leben lang abhängig von Transfusionen.
Über die Risiken einer Bluttransfusion klärt der transfundierende Arzt auf. Das Risiko der Übertragung von Viruserkrankungen (Hepatitis B, Hepatitis C, HIV) durch eine Bluttransfusion ist heutzutage aufgrund von umfangreichen Untersuchungen von Spendern und Blutprodukten außerordentlich gering.
Während regelmäßiger Bluttransfusionen sind verschiedene Untersuchungen zur Überwachung der Therapie notwendig (s. Downloadbereich).
Steroide
Steroide, auch Glukokortikoide genannt, sind körpereigene Hormone (Botenstoffe), die die Blutbildung anregen können. Der genaue Wirkmechanismus von Steroiden auf die Blutbildung ist noch unklar. Zur Behandlung der DBA werden künstlich hergestellte Steroide eingesetzt. Da die Substanzen jedoch auch wachstumshemmend wirken, sollten sie bei Kindern vor dem ersten Geburtstag möglichst vermieden werden. Die Dosis der Steroide wird grundsätzlich auf das Körpergewicht der Patient*innen bezogen.
Bei Säuglingen mit DBA, die Transfusionen benötigen, wird nach dem ersten Geburtstag ein sogenannter erster Steroidversuch empfohlen. Meist wird ca. 2 Wochen nach einer Transfusion mit einer Steroidbehandlung in einer Tagesdosis von 2 mg/kg Körpergewicht begonnen (bei Erwachsenen beträgt die Maximaldosis 60 mg). Das Blutbild wird dabei regelmäßig überprüft. Steigt der Hämoglobinwert an oder hält sich (ohne Transfusion) stabil, wird die Steroid-Dosis schrittweise verringert. Langfristig wird die niedrigste Dosis gesucht, die ausreicht, um den Hämoglobinwert im Normbereich zu halten.
Sollte vier Wochen nach Beginn eines Steroidversuchs der Hämoglobinwert deutlich abgefallen sein, war der Behandlungsversuch nicht erfolgreich und es wird zunächst wieder eine Therapie mit regelmäßigen Transfusionen aufgenommen. Ein zweiter (und in der Regel letzter) Behandlungsversuch mit Steroiden wird dann meist 1 Jahr später durchgeführt.
Eine Behandlung mit Steroiden geht je nach Höhe der Dosis mit akuten und chronischen Nebenwirkungen einher. Akute Nebenwirkungen wie Appetitzunahme, Gewichtszunahme, Veränderungen der Gesichtsform, Stimmungsschwankungen, gelegentlich auch erhöhter Blutdruck und Blutzucker werden bei hohen Dosen (2 mg/kg Körpergewicht) häufig beobachtet; sie bilden sich unter Dosisreduktion rasch wieder zurück. Chronische Nebenwirkungen treten in der Regel nur bei zu hohen Dosierungen (Grenze ca. 0,2 - 0,3 mg/kg Gewicht/Tag) auf und umfassen unter anderem Störungen des Blutzuckerhaushalts, Veränderungen des Körperbaus und der Haut, einen hohen Blutdruck, Sehstörungen, Wachstumsverzögerungen, Knochenschwäche und Magenschleimhautentzündungen. Eine langfristige Behandlung mit Steroiden sollte daher eine Dosierung von 0,2 - 0,3 mg pro kg Körpergewicht nicht überschreiten. Auch unter einer Therapie mit Steroiden sind regelmäßige Verlaufsuntersuchungen notwendig (s. Downloadbereich).
Allogene Stammzelltransplantation
Eine allogene Stammzelltransplantation (SZT) ist momentan die einzige Therapie, die die Erkrankung im Knochenmark heilt und damit eine endgültige Behandlung der Anämie darstellt. Als Quelle für die SZT empfehlen wir die Nutzung von Knochenmark-Stammzellen bei Kindern und Jugendlichen, periphere Blut-Stammzellen stellen eine nachrangige Möglichkeit dar. Da es bei einer SZT zu lebensbedrohlichen und langfristig schweren Nebenwirkungen kommen kann, müssen die Risiken und der Nutzen der Behandlung für jeden Patienten individuell abgewogen werden. Die Chance einer Heilung der Anämie durch die SZT bei optimaler Vorbereitung und Auswahl der Patient*innen ist mittlerweile sehr hoch. Das Risiko von schweren und tödlichen Komplikationen einer SZT steigt bei DBA-Patient*innen mit dem Alter rasch an, so dass die SZT meist nur transfusionsbedürftigen Kindern unter 10 Jahren als Behandlungsoption angeboten wird, wenn ein optimaler Spender (Geschwisterspender oder passender Fremdspender) zur Verfügung steht. Ein Aufklärungsgespräch zur SZT bieten wir allen Familien nach einem erfolglosen ersten Steroidversuch gerne an.
Bei schweren Komplikationen der Transfusionsbehandlung der DBA, wie z. B. bei einer Entwicklung von Antikörpern gegen Erythrozyten (Alloimmunisierung), die eine Fortführung von Bluttransfusionen unmöglich machen, oder nicht beherrschbaren Nebenwirkungen von Medikamenten zum Eisenentzug, kann eine SZT auch bei älteren DBA-Patient*innen notwendig werden. Ebenso kann ein dauerhafter Abfall der Leukozyten (führt zu Infektionen) oder der Thrombozyten (können nicht dauerhaft durch Transfusionen ersetzt werden) ein Grund für eine SZT sein. Bei älteren Kindern (> 10 Jahre) und Erwachsenen sollten die Indikation für eine SZT nur von einem sehr erfahrenen Behandlungsteam überprüft werden.
Gentherapie
Eine Gentherapie wird heute bereits für eine andere Knochenmarkserkrankung, die Fanconi-Anämie, in Studien erfolgreich eingesetzt. Auch für DBA werden voraussichtlich in wenigen Jahren gentherapeutische Behandlungen angeboten werden können. Bei einer Gentherapie müssen vom Patienten zunächst frische Stammzellen der Blutbildung gesammelt werden. Hierzu werden die Stammzellen mit einem Medikament aus dem Knochenmark der Patient*innen in das Blut gelockt und das Blut außerhalb des Körpers apparativ in einer Leukapherese-Einheit (vom Prinzip her ähnlich einer Blutwäsche) in seine Bestandteile aufgetrennt. In die so gewonnenen Stammzellen wird über bestimmte Transportvehikel Vektoren) ein gesundes Gen als Ersatz für das jeweils defekte Gen eingebracht (Transduktion) oder das defekte Gen korrigiert (Genome Editing). Dadurch kann der Gendefekt beseitigt und die Funktionsstörung der Ribosomen behoben werden. Zurzeit werden Gentherapien für das Gen RPS19 entwickelt.
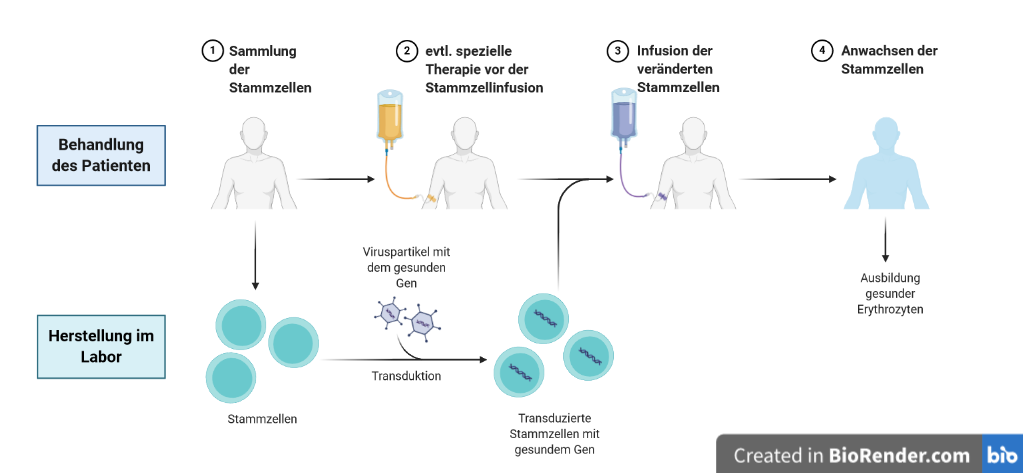
Abbildung 4: Schema einer Gentherapie
Bisher für eine Behandlungsempfehlung nicht ausreichend durch Studien belegte, individuelle, experimentelle Therapieoptionen
Bei DBA-Patient*innen wurden eine ganze Reihe anderer Medikamente zur Therapie ausprobiert, u.a. intravenöses Immunglobulin, Erythropoeitin (EPO), Interleukin-3, Androgene, Ciclosporin A, Antithymozytenglobulin (ATG) und Metoclopramid (oft verwendetes Medikament gegen Übelkeit). Alle diese Therapieversuche waren nicht erfolgreich. Mit Metoclopramid konnten sehr geringe Ansprechraten in Form eines minimalen Anstiegs des Hämoglobinwertes von DBA-Patient*innen erreicht werden. Die Wirkung wird über eine Beeinflussung des Hormons Prolaktin vermittelt.
Eine bestimmte Aminosäure, L-Leucin, wurde in einer größeren Studie in den USA zur Behandlung von DBA-Patient*innen eingesetzt, nachdem in Einzelfällen nachvollziehbare hämatologische Remissionen einer DBA berichtet wurden. Die Studie erbrachte jedoch nur sehr geringe Ansprechraten, unter 10% der behandelten Patient*innen zeigten Anstiege des Hämoglobinwerts. Ein Teil der Kinder zeigte während der Behandlungsphase eine etwas verbesserte Wachstumsgeschwindigkeit.
In letzter Zeit wurden einzelne Fallberichte bekannt, bei denen DBA-Patient*innen mit Elthrombopag behandelt wurden, einem Thrombopoeitin-Rezeptor-Agonisten, der für die Behandlung von Patient*innen mit einer immunvermittelten Thrombozytopenie entwickelt wurde. Diese Patient*innen erreichten durch eine oftmals sehr hoch dosierte Behandlung mit Elthrombopag eine Unabhängigkeit von Bluttransfusionen. Studien, die diesen Effekt in einer großen Anzahl von Patient*innen bestätigen und v.a. auf Nebenwirkungen untersuchen, fehlen hierzu allerdings.
Momentan wird nach weiteren Medikamenten für die Behandlung der DBA geforscht.
Fehlbildungen
Viele der angeborenen Fehlbildungen (wie Daumenfehlbildungen oder Fehlbildungen des Unterarmknochens) können durch bestimmte Operationsverfahren korrigiert werden. Auch Fehlbildungen an den inneren Organen (beispielsweise der Harnwege, im Magen-Darm-Trakt oder am Herzen) können operativ behandelt werden. Das interdisziplinäre Behandlungsteam wird gemeinsam mit den entsprechenden Fachdisziplinen aus der Chirurgie und Kardiologie, Nephrologie oder Gastroenterologie individuell darüber informieren, welche Eingriffe sinnvoll und angezeigt sind.
Bei DBA-Patient*innen können im Verlauf sowohl Komplikationen der Erkrankung selbst als auch Komplikationen der Behandlung auftreten. Im Rahmen einer Transfusionstherapie kommt es ohne Eisenentzugstherapie zu einer schweren Eisenüberladung, die sich in Form von Herzerkrankungen (z.B. Herzschwäche, Herzrhythmusstörungen), Lebererkrankungen (z.B. Leberentzündung, Gelbsucht, Bauchwassersucht, Milzvergrößerung, Blutungen, Leistungsdefizit, Konzentrationsstörung), Erkrankungen der Hormondrüsen (z.B. Diabetes mellitus, Kleinwuchs, verzögerte Pubertät, Entwicklungsstörung, Müdigkeit) und einer vermehrten Knochenbrüchigkeit (Osteoporose) zeigen kann. Diese Komplikationen können mit einer frühzeitigen und gut gesteuerten Eisenentzugstherapie vermieden werden.
Selten kann es durch zahlreiche Transfusionen zu einer sog. Alloimmunisierung kommen. Hierbei erkennt das Immunsystem der Patient*innen die fremden Eiweiße auf der Hülle der Erythrozyten des Blutspenders und bildet Antikörper gegen sie. In der Folge kann eine nächste Transfusion vom gleichen oder ähnlichen Spendern mit den gleichen fremden Eiweißen zu einer schweren Transfusionsreaktion führen. Dabei werden die Erythrozyten zerstört und es kann zu Thrombosen (Blutgerinnseln) kommen. Es gibt Situationen, in denen für DBA-Patient*innen mit Alloimmunisierung immer weniger passende>Erythrozytenkonzentrate gefunden werden können und letztlich nur noch eine Stammzelltransplantation als Therapieoption übrig bleibt.
Der DBA liegt eine Bildungsstörung von Blutzellen im Knochenmark zugrunde. In wenigen Fällen ist die Bildungsstörung nicht auf die Erythrozyten beschränkt, sondern betrifft fortschreitend auch Leukozyten und Thrombozyten. Bei dieser sogenannten Panzytopenie kann eine Verringerung der Zahl der Leukozyten zu Fieber, Aphten im Mund und schweren Infektionen führen, und eine niedrige Zahl von Thrombozyten zu spontanen Haut- und Schleimhautblutungen wie Nasenbluten. Auch in diesen Fällen kann eine SZT indiziert sein.
Die DBA wird mittlerweile auch als Tumorprädispositionssyndrom verstanden, d.h. als eine Erkrankung, bei der bestimmte Krebserkrankungen häufiger sind bzw. früher auftreten als bei nicht betroffenen Menschen. Die genaue Art der Krebserkrankungen, die vermehrt auftreten und die Höhe des zusätzlichen Risikos für Krebserkrankungen sind noch nicht bekannt, zu den gehäuft auftretenden Tumoren gehören aber Brustkrebs, Darmkrebs, Knochentumoren und Hautkrebs. DBA-Patient*innen wird daher empfohlen, eine jährliche ärztliche Untersuchung durchführen zu lassen, an den gesetzlichen Früherkennungsprogrammen teilzunehmen und bei Symptomen kurzfristig einen Arzt aufzusuchen. Eine spezielle Empfehlung für Früherkennungsuntersuchungen bei DBA-Patient*innen kann zurzeit noch nicht ausgesprochen werden.
Wenige DBA-Patient*innen zeigen direkt nach Geburt oder im Laufe des Lebens, vor allem im Erwachsenenalter, Anzeichen für eine Schwäche des Immunsystems. Hierbei kann es zum Auftreten von gehäuften Infektionen, zum Auftreten von schweren Infektionen durch Bakterien, zum Auftreten von Infektionen durch sogenannte untypische Erreger und zum Auftreten langanhaltender oder untypisch verlaufender Infektionen kommen. Es wird angeraten, dass DBA-Patient*innen alle empfohlenen Impfungen inkl. der Grippeschutzimpfung erhalten.
Bei älteren erwachsenen Patient*innen kann es aus nicht ausreichend verstandenen Ursachen zu Einbußen an körperlicher und mentaler Leistungsfähigkeit kommen. Patient*innen berichten, dass die Belastbarkeit im Sport im Vergleich zu Gleichaltrigen abnimmt und Konzentrations- und Gedächtnisprobleme früher als erwartet auftreten. Eine Datensammlung und Befragung erwachsener DBA-Patient*innen zu dieser sehr wichtigen Beobachtung findet aktuell über ein Selbsteingabemodul statt.
Mit jeder Bluttransfusion wird den betroffenen Patient*innen neben den dringend benötigten Erythrozyten auch das hierin gebundene Eisen übertragen. Jedes Erythrozytenkonzentrat enthält ungefähr 200 – 250 mg Eisen, am Tag nimmt ein Mensch bei ausgewogener Ernährung nur ca. 1 – 2 mg Eisen zu sich. Ab einer Transfusionsmenge von ca. 10 – 15 Erythrozytenkonzentraten kommt es zu einer Eisenüberladung. Zusätzlich nehmen Patient*innen mit einer Anämie dauerhaft zu viel Eisen über den Darm auf.
Bei einer Eisenüberladung kann Eisen nicht mehr an seine eigentlichen Transportproteine gebunden werden und lagert sich in verschiedenen Organen ab, vor allem im Herz, der Leber und den hormonproduzierenden Drüsen. Hier kann es schwere Organschäden hervorrufen, eine Schädigung des Herzens ist die häufigste Todesursache bei Patient*innen mit angeborenen Anämien. Die frühzeitige Diagnose und Messung der Eisenlast hat große Bedeutung für die Prognose der betroffenen Patient*innen.
Im Blut lässt sich die Eisenbeladung mittels der Bestimmung der Transferrinsättigung und des Ferritinwerts messen. Eine erhöhte Sättigung des Transportproteins Transferrin mit Eisenmolekülen zeigt eine Eisenüberladung an. Der Ferritinwert zeigt an, wie viel Speichereisen im Körper in Organen, vor allem in der Leber, vorhanden ist. Der Ferritinwert steigt aber auch bei Entzündungen oder Infektionen an und fungiert daher eher als Verlaufsparameter, der die Ausprägung der Eisenüberladung nur ungenau angeben kann.
Eine wesentliche genauere Messung der Eisenüberladung ist mittels Magnetresonanztomographie (MRT) der betroffenen Organe Leber, Pankreas und Herz möglich. Das MRT-Gerät misst den Einfluss des gespeicherten Eisens auf die Darstellung der Organe; im erzeugten MRT-Bild erscheint eine eisenüberladene Leber deutlich dunkler als eine gesunde Leber (s. Abbildung 5).
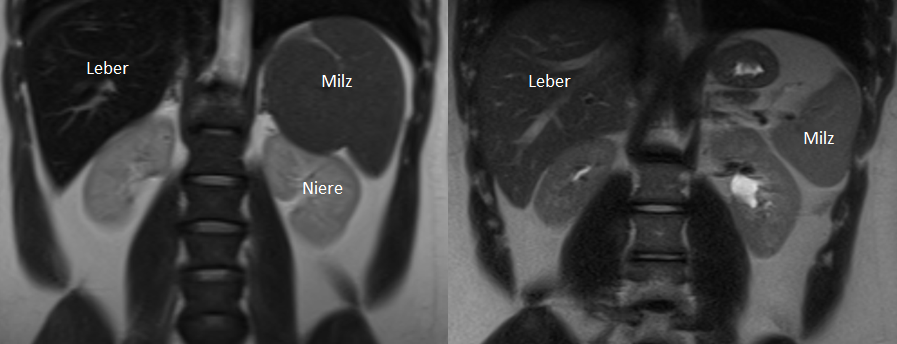
Abbildung 5: MRT eines polytransfundierten Patienten mit „schwarzer“ Leber und Milz (links) sowie eines gesunden Menschen mit „hellen“ Organen (rechts).
Dabei spielt die Wechselwirkung zwischen dem magnetischen Eisen und dem Magnetfeld des MRT, das für die Bilderzeugung notwendig ist, eine wichtige Rolle. Eine MRT-Untersuchung ist schmerzlos und birgt keine Strahlenbelastung. Die MRT-Untersuchung zur Bestimmung der Eisenüberladung sollte in einem der großen Behandlungszentren für Blutkrankheiten durchgeführt werden, da die Untersuchungsergebnisse interdisziplinär von einem Team aus Spezialisten für (Kinder-) Blutkrankheiten und (Kinder-)Radiologen bewertet werden müssen.
Mit einer Entnahme (Biopsie) von Lebergewebe, kann man die Eisenkonzentration im Lebergewebe auch direkt bestimmen. Hierzu wird unter einer Kontrolle mit Ultraschall eine spezielle Nadel durch die Bauchdecke der Patient*innen in die Leber geführt und eine kleine Menge an Lebergewebe entnommen. Dabei kann es zu Komplikationen wie Blutungen, Infektionen, Leckage von Galleflüssigkeit oder Entzündungen kommen. Bei Kindern und Jugendlichen wird der Eingriff in Narkose durchgeführt und erfordert eine stationäre Überwachung. Zudem besteht das Risiko, dass das die Eisenüberladung in dem punktierten Leberareal nicht die Eisenüberladung der ganzen Leber widerspiegelt, da die Eisenbeladung der Leber oft fleckförmig ist. Heutzutage wird im Allgemeinen eine Lebereisenmessung durch die MRT angestrebt und nur bei zusätzlichen Fragen (z. B. Leberentzündung, Leberzirrhose) eine Leberbiopsie durchgeführt.
Eine Messung der Organeisenkonzentration mittels SQUID (superconducting quantum interference device), mit der lange Zeit sehr genaue Werte für die Organeisenkonzentration bestimmt werden konnten, ist seit kurzer Zeit in Deutschland nicht mehr verfügbar.
Die Behandlung von Patient*innen mit seltenen Anämien sollte durch ein erfahrenes Behandlungszentrum für Blutkrankheiten erfolgen. Hier sollten regelmäßige Untersuchungen zur Messung der Eisenüberladung und Suche nach möglichen Komplikationen der Eisenüberladung erfolgen. Je frühzeitiger eine Komplikation entdeckt wird, desto besser ist sie behandelbar. Die für DBA-Patient*innen empfohlenen regelmäßigen Untersuchungen haben wir in einer Checkliste zusammengestellt (siehe Downloadbereich).
Der menschliche Organismus kann überschüssiges Eisen nicht selbstständig ausscheiden. Deshalb ist eine Behandlung mit Medikamenten, mit deren Hilfe das Eisen aus dem Körper ausgeschleust werden kann, notwendig (Eisenentzugstherapie). Hierbei handelt es sich um Substanzen, die mit dem überschüssigen Eisen eine feste Verbindung eingehen, ein so genanntes Chelat bilden (Chelatbildner, Chelatoren). So gebunden kann das überschüssige Eisen über den Urin und/oder den Stuhl ausgeschieden werden. Die Behandlung sollte ab einem bestimmten Grenzwert für das Ferritin (meist 2x Messung mit einem Wert >1000 ng/ml), einem bestimmten Wert für die Eisenbeladung im Lebergewebe (meist >4,5 mg Eisen/g Lebertrockengewicht) oder ab ca. 10 – 15 Transfusionen beginnen. Der Beginn einer Eisenentzugstherapie wird international aber nicht vor dem 2. Geburtstag empfohlen.
Derzeit kommen drei verschiedene Eisenchelatbildner zum Einsatz: Deferoxamin (DFO, Handelsname Desferal ®), Deferasirox (DSX, Handelsname Exjade ®) und Deferipron (DFP, Handelsname Ferriprox ®). Deferoxamin wird mit verschiedenen Techniken über die Haut (subkutan), über einen Muskel (intramuskulär) oder über eine Vene (intravenös) verabreicht. Deferasirox kann von den Patient*innen als Tablette oder zermörsertes Pulver geschluckt werden (oraler Chelatbildner). Für einige Patient*innen mit DBA, für die die Behandlung mit Deferoxamin oder Deferasirox nicht in Frage kommt, kann der orale Chelatbildner Deferipron zur Verfügung stehen. Der Einsatz der verschiedenen Chelatoren ist abhängig vom Alter, den individuellen Nebenwirkungen und natürlich von der Wirksamkeit der Medikamente auf die Eisenüberladung in den verschiedenen Organen. Bei Patient*innen mit schwerster Eisenüberladung und Herzversagen, Herzrhythmusstörungen oder Diabetes mellitus können sehr intensive Maßnahmen zum Eisenentzug erforderlich sein. Zusammenfassend sollte eine Eisenentzugstherapie nur vom Spezialisten für Blutkrankheiten gesteuert und überwacht werden. Die unter einer Eisenentzugstherapie empfohlenen zusätzlichen regelmäßigen Untersuchungen haben wir in einer Checkliste im Downloadbereich hinterlegt.
Die Prognose von Patient*innen mit DBA ist ganz wesentlich von der Qualität der medizinischen Versorgung mitbestimmt. Dabei spielen altersentsprechende Aufklärungen und die Möglichkeit zur Mitarbeit seitens der betroffenen Patient*innen und ihrer Angehörigen eine wichtige Rolle.
Fast alle Patient*innen mit einer Diamond-Blackfan-Anämie (DBA) können einen Kindergarten und die Schule erfolgreich besuchen, eine Ausbildung oder ein Studium durchführen, einen Beruf erlernen und ausüben. Bei manchen Patient*innen (20-30%) kommt es im Verlauf des Lebens sogar zu einer sogenannten Remission, d.h. zum spontanen Verschwinden der Anämie. Die Mehrzahl der Patient*innen bleibt aber langfristig auf Steroide oder Transfusionen angewiesen. Die Langzeitprognose von DBA-Patient*innen kann durch die Komplikationen der Transfusionstherapie oder einer zu hoch dosierten Steroid-Therapie eingeschränkt sein. Die Rolle des erhöhten Krebsrisikos auf das Langzeitüberleben lässt sich zurzeit noch nicht abschätzen.
Der Verlauf der DBA bei einzelnen Patient*innen lässt sich bisher nicht voraussagen. Eine DBA kann selbst unter günstigsten beziehungsweise ungünstigsten Voraussetzungen ganz unerwartet verlaufen. Die allgemeine Lebenserwartung von DBA-Patient*innen heute ist noch nicht bekannt. Zum einen ist die Erkrankung erst vor 70 Jahren beschrieben worden, so dass es noch nicht viele Patient*innen gibt, die über einen langen Zeitraum beobachtet wurden. Zum anderen wurde die Therapie in den letzten Jahrzehnten so stark optimiert (insbesondere die Therapie der Eisenüberladung), dass sich die Prognose aller angeborenen Anämien deutlich verbessert hat.
Da die DBA sehr komplexe Erkrankung ist, gibt es zahlreiche Forschungsfragen zu beantworten. In zahlreichen Registern für DBA-Patient*innen weltweit wird der natürliche Verlauf der DBA erfasst. Insbesondere die Erfassung des Verlaufs von erwachsenen Patient*innen, deren Gesundheits- und Versorgungszustand, sowie die Erfassung von Komorbiditäten wie auch Krebserkrankungen im späteren Lebensalter ist ein Kernpunkt unserer aktuellen Forschung. Dazu entwickeln wir gerade ein Eingabemodul, in das erwachsene Patient*innen ihre Krankheitsdaten online selbstständig eingeben können. Wir lernen aktuell von den älteren DBA-Patient*innen, um die Therapie junger Patient*innen zu verbessern.
Auch wenn bei 70-80% der Patient*innen, die klinisch eine DBA haben, eine krankheitsverursachende genetische Veränderung (pathogene genetische Variante) gefunden werden kann, bleibt noch ein relevanter Anteil an Patient*innen übrig, bei denen die entsprechende Veränderung noch gefunden werden muss. Die Identifikation neuer DBA-verursachender pathogener genetischer Varianten wird ergänzt durch die Entwicklung neuer funktioneller Untersuchungen, damit auch bei fehlendem Mutationsnachweis die Diagnose der DBA gestellt werden kann. Eine mögliche, aber noch nicht für die Routinediagnostik einsetzbare Methode ist die Darstellung der Profile der Ribosomen eines Menschen. Diese Ribosomenprofile sind bei Patient*innen mit DBA auffällig und unterscheiden sich von gesunden Menschen.
Um zu verstehen, wie die Erkrankung der Ribosomen zu einer gestörten Blutbildung führt und warum eine Steroidtherapie in manchen, aber nicht allen Patient*innen zu einer Remission der Anämie führt, wird in zahlreichen Laboren weltweit die Pathogenese der DBA untersucht. Eine ganz besonders wichtige Entdeckung war, dass der für die rote Blutreihe essentielle Transkriptionsfaktor GATA1 durch die Veränderung der Ribosomen von DBA-Patient*innen nicht mehr richtig hergestellt werden kann. Interessanterweise haben Patienten mit einer angeborenen GATA1-Mutation eine Erkrankung, die der der klassischen DBA sehr ähnlich ist.
Da die allogene Stammzelltransplantation weiterhin die einzige kurative Therapieoption für DBA-Patient*innen darstellt und damit nur die hämatologischen Probleme, nicht aber andere Organschäden behandelt werden können, wird intensiv nach weiteren Medikamenten für die Behandlung geforscht. Hierzu existieren große, informationstechnologisch sehr herausfordernde Plattformen, in denen nach Molekülen gesucht wird, die die Funktion von Ribosomen wiederherstellen können. Außerdem werden bereits für andere Erkrankungen zugelassene Medikamente in Form von klinischen Studien an DBA-Patient*innen getestet, u.a. Elthrombopag in einer Studie in den USA.
Im Bereich der allogenen Stammzelltransplantation wird in klinischen Studien nach dem besten Zeitpunkt und der besten Form der Konditionierung, also der vorbereitenden Chemotherapie vor einer SZT, für DBA-Patient*innen gesucht.
Wie bereits im Abschnitt „Behandlungsmethoden einer DBA“ erwähnt, wird momentan die Möglichkeit einer Gentherapie bei DBA-Patient*innen mit einer pathogenen genetischen Veränderung im RPS19-Gen entwickelt. Hierzu sind zunächst Versuche im Reagenzglas notwendig, auch wenn das Prinzip einer Gentherapie bei angeborenen Anämien in Form der Fanconi-Anämie, einer Erkrankung, die auch mit Fehlbildungen und einem Versagen des Knochenmarks einhergehen kann, bereits erfolgreich getestet wurde.